SOFTWARE & DATABASE
JOB (Japan Omics Browser)
- Takahashi Y et al. (2025) JOB: Japan Omics Browser provides integrative visualization of multi-omics data. BMC Genomics 26:451. [Pubmed]
- Wang QS et al. (2024) Statistically and functionally fine-mapped blood e/pQTLs from 1,405 humans reveal their distinct regulation patterns and disease relevance. Nat Genet 56:2054-2067. [Pubmed]

- web page [Link]
HTE-PRS (Herogenous Treatment Effect across Polygenic Risk Score)
- Naito T, Inoue K et al. (2024) Machine learning reveals heterogeneous associations between environmental factors and cardiometabolic diseases across polygenic risk scores. Commun Med 4:181. [Pubmed]

- github page [Link]
scLinaX (single-cell Level inactivated X chromosome mapping)
- Tomofuji Y et al. (2024) Quantification of the escape from X chromosome inactivation with the million cell-scale human single-cell omics datasets reveals heterogeneity of escape across cell types and tissues. Cell Genom 100625. [Pubmed]

- github page [Link]
The KFc (Knockoff-FINMAP combination) method
- Wang QS et al. (2023) Estimating gene-level false discovery probability improves eQTL statistical fine-mapping precision. NAR Genom Bioinform 5:lqad090. [Pubmed]
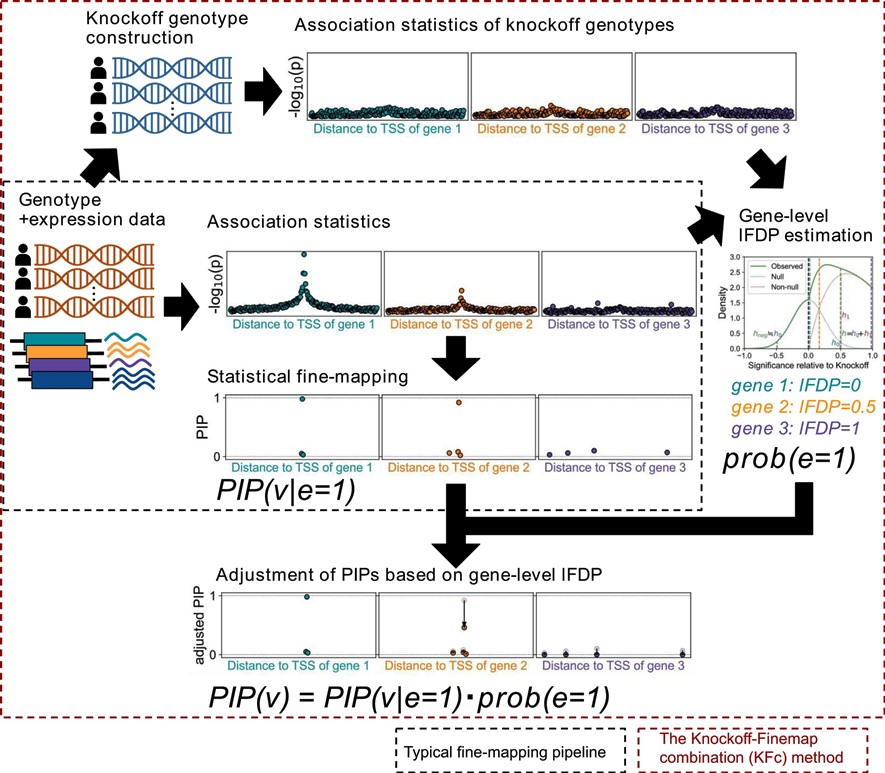
- github page [Link]
JMAG (Japanese Metagenome Assembled Genomes Platform)
JVD (Japanese Virus Database)
- Tomofuji Y et al. (2022) Prokaryotic and viral genomes recovered from 787 Japanese gut metagenomes revealed microbial features linked to diets, populations, and diseases. Cell Genom 2:100219 [Pubmed]
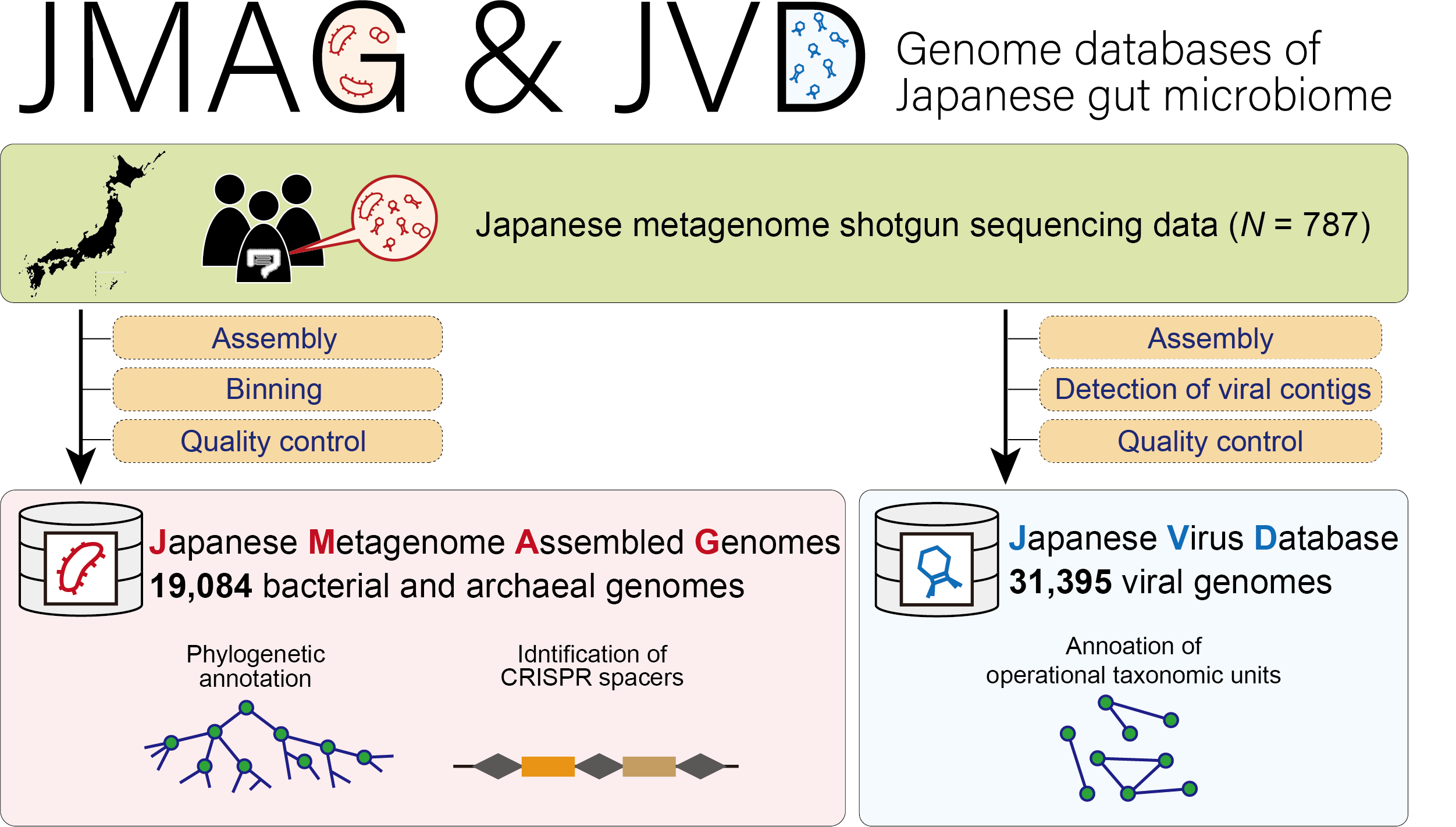
- JMAG: [Link], JVD: [Link], CRIPR spacer: [Link]
KIRAP (Killer Immunoglobulin-like Receptor variant Analytical Platform)
- Sakaue S et al. (2022) Decoding the diversity of killer immunoglobulin-like receptors by deep sequencing and a high-resolution imputation method. Cell Genom 2:100101. [Pubmed]
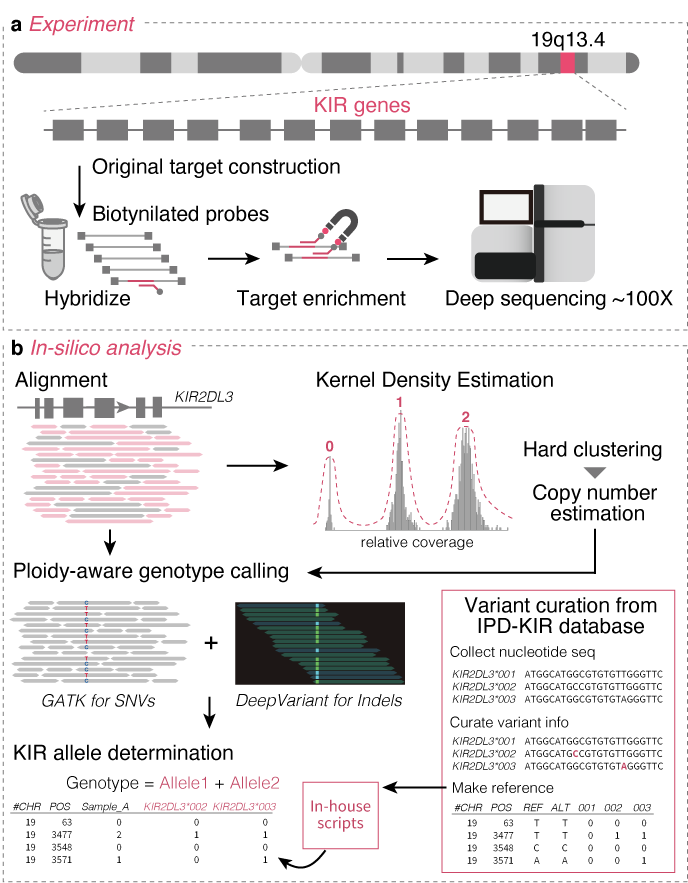
- github page [Link]
OMARU (Omnibus Metagenome-wide Association study with RobUstness)
- Kishikawa T et al. (2022) OMARU: a robust and multifaceted pipeline for metagenome-wide association study. NAR Genom Bioinform 4:lqac019. [Pubmed]
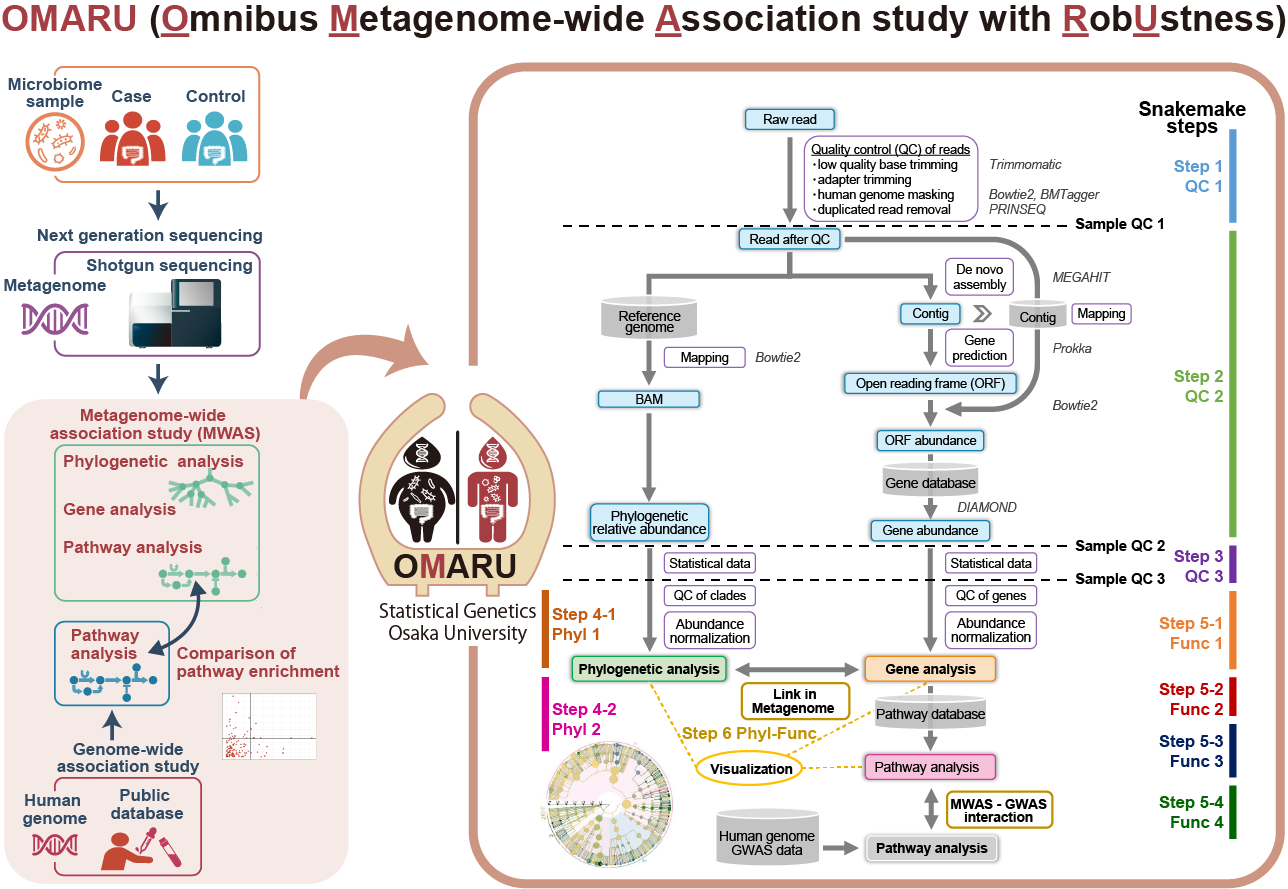
- github page [Link]
PheWeb.jp (GWAS database of Biobank Japan and cross-population studies)
- Sakaue S, Kanai M et al. (2021) A global atlas of genetic associations of 220 deep phenotypes. Nat Genet 53:1415-1424. [Pubmed]
- web page [Link]
Trans-Phar (Integration of TWAS and pharmacological database)
- Konuma T, Ogawa K, Okada Y. (2021) Integration of genetically regulated gene expression and pharmacological library provides therapeutic drug candidates. Hum Mol Genet 30:294-304. [Pubmed]
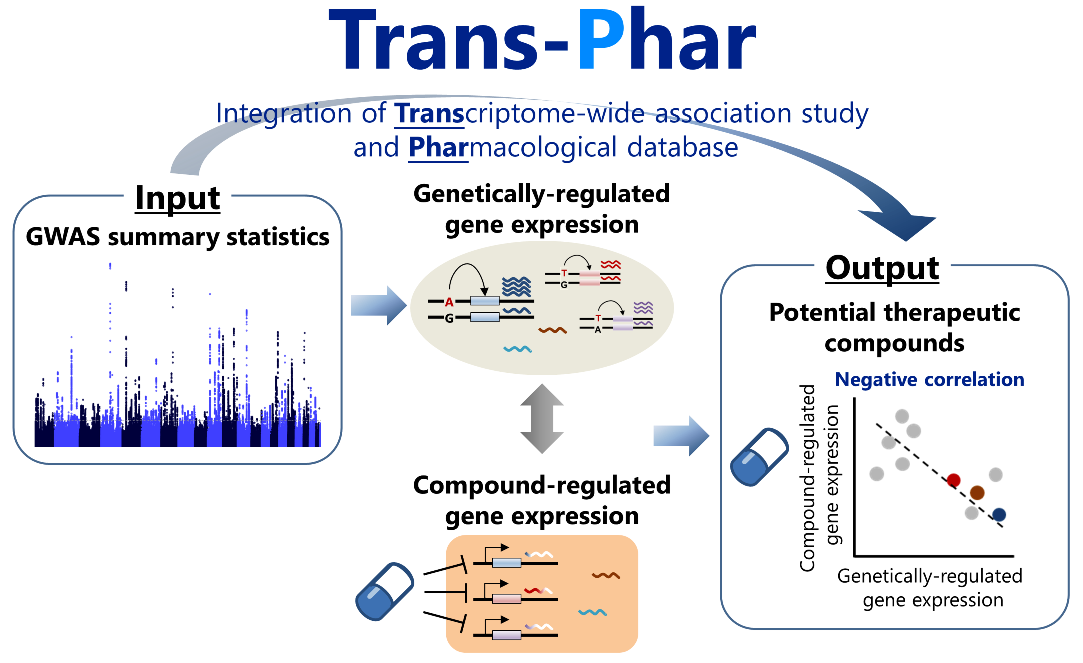
- github page [Link]
DEEP*HLA (DEEP learning for HLA allelic imputation)
- Naito T et al. (2021) A deep learning method for HLA imputation and trans-ethnic MHC fine-mapping of type 1 diabetes. Nat Commun 12:1639. [Pubmed]
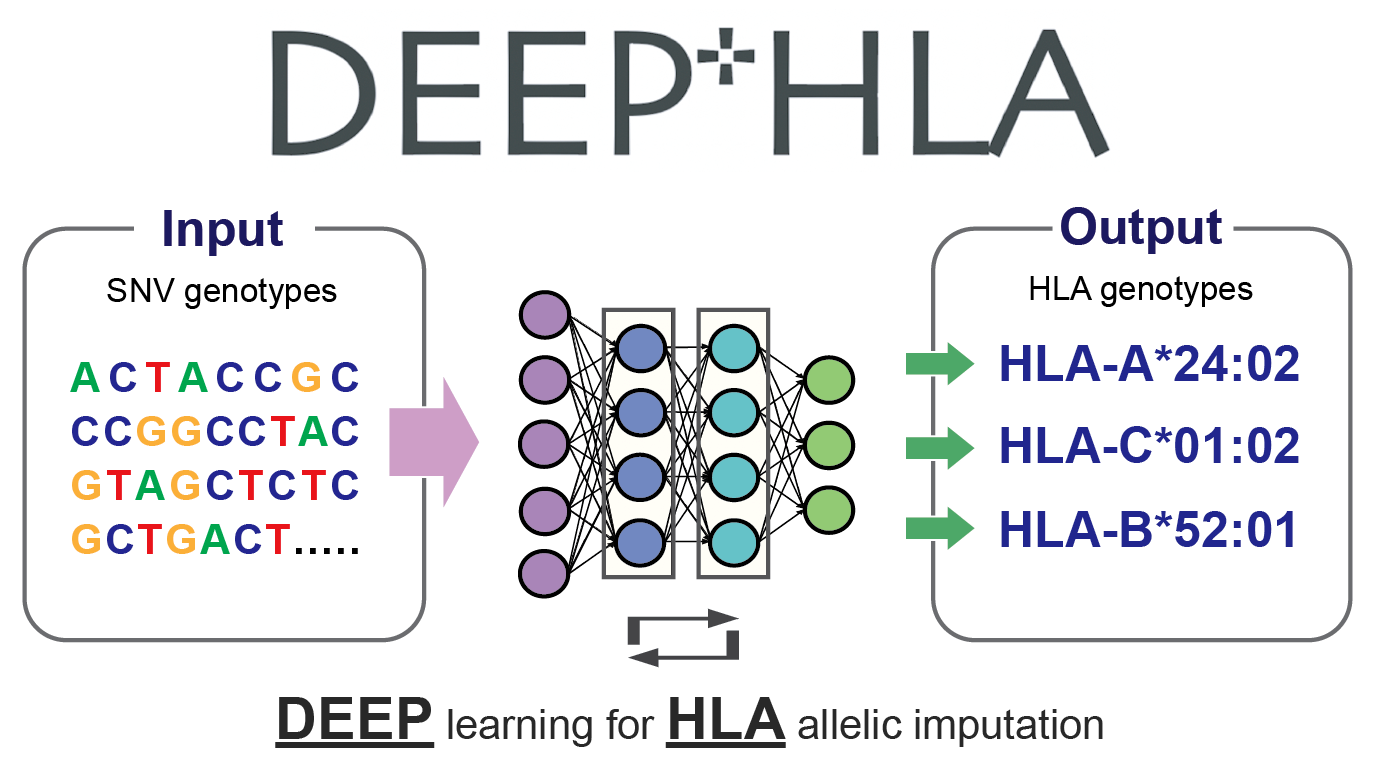
- github page [Link]
Obelisc (Observational linkage scan)
- Sonehara K and Okada Y. (2021) Obelisc: an identical-by-descent mapping tool based on SNP streak. Bioinformatics 36:5567-5570. [Pubmed]
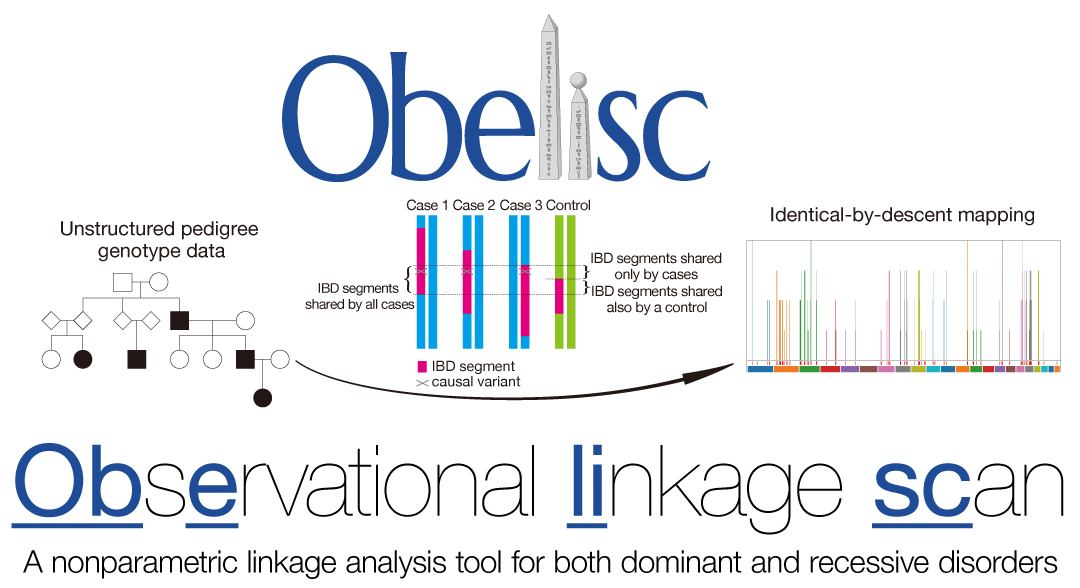
- github page [Link]
GREP (Genome for REPositioning drugs)
- Sakaue S and Okada Y. (2019) GREP: Genome for REPositioning drugs. Bioinformatics 35:3821-3823. [PubMed]
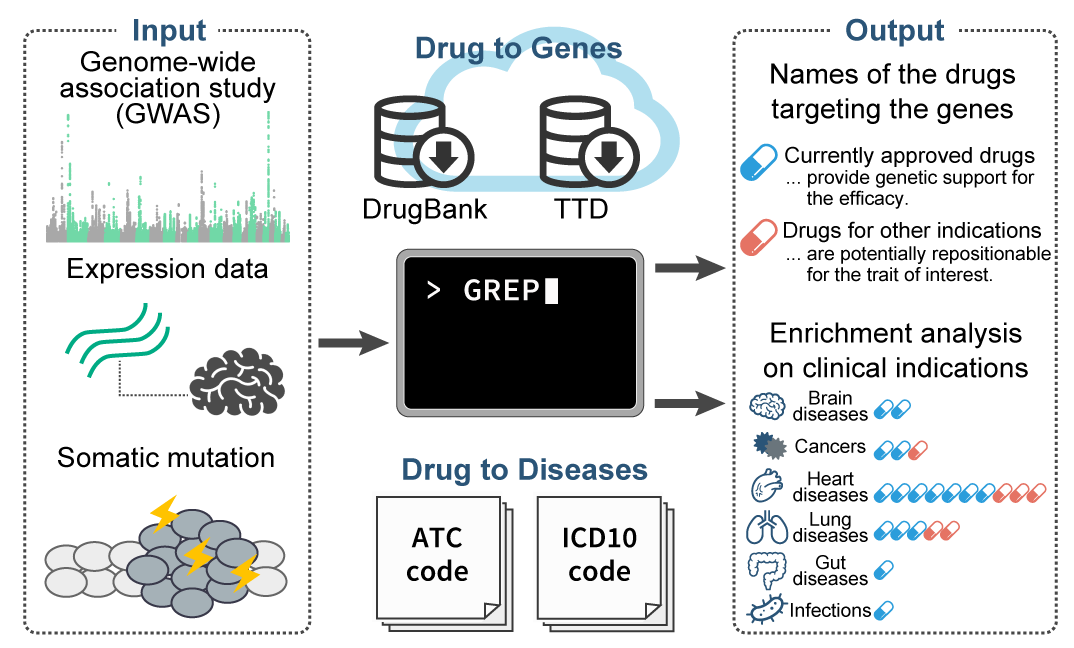
- github page [Link]
MIGWAS (miRNA-target gene networks enrichment on GWAS)
- Sakaue S et al. (2018) Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res 46:11898-11909. [PubMed]
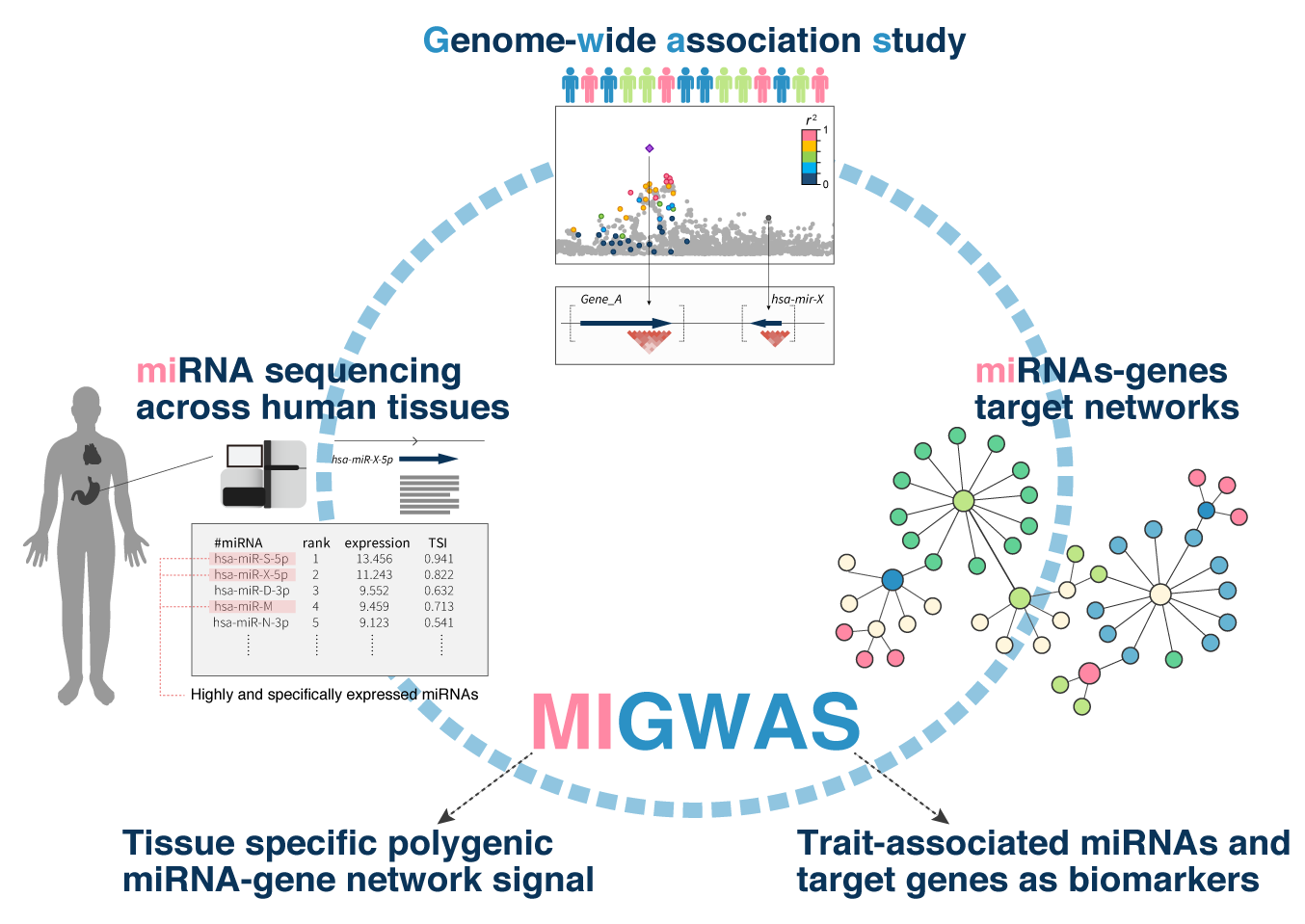
- github page [Link]
eLD (entropy-based Linkage Disequilibrium index between multiallelic sites)
- Okada Y. (2018) eLD: entropy-based Linkage Disequilibrium index between multi-allelic sites. Hum Genome Var 5:29. [PubMed]
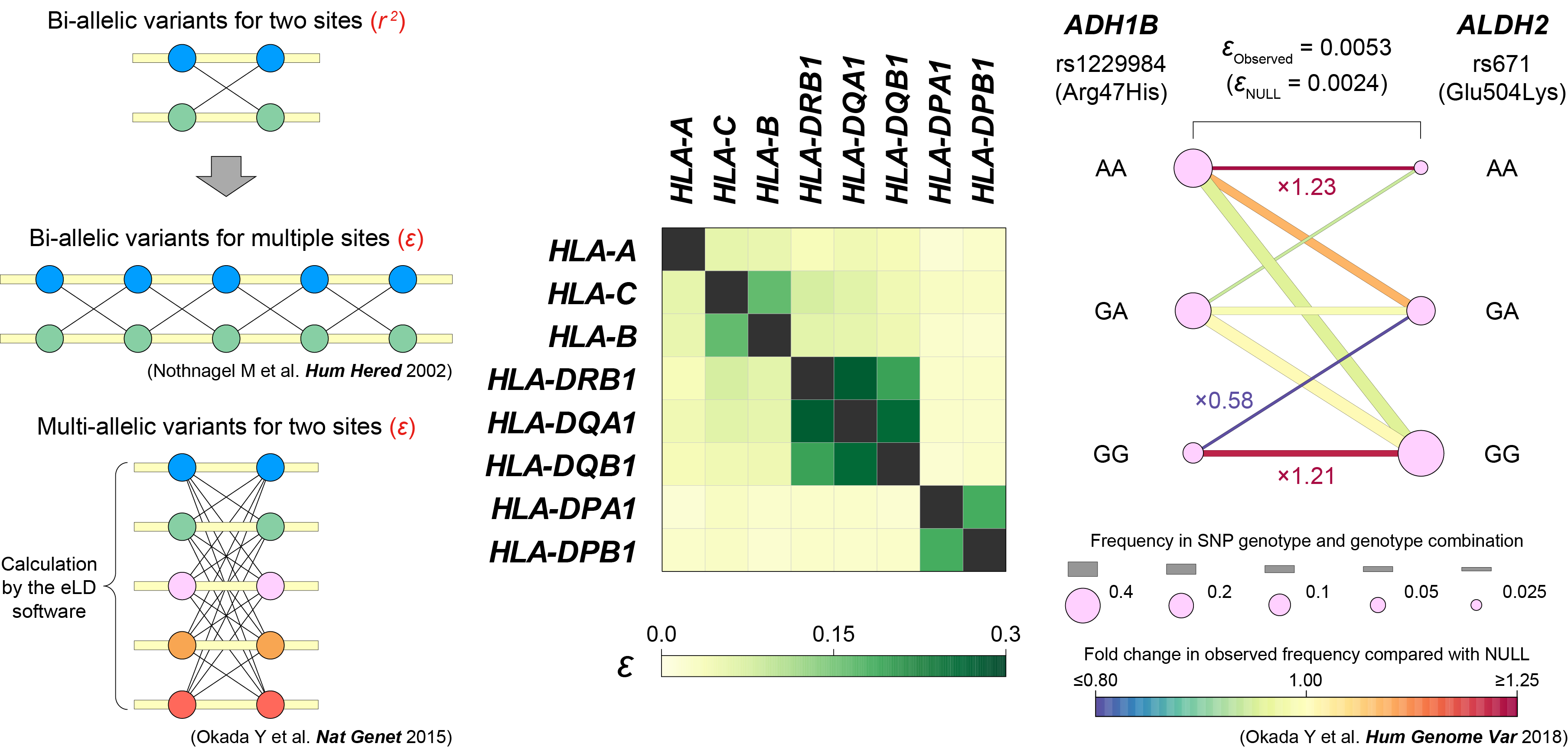
- R script [Link]
Grimon (Graphical interface to visualize multi-layer omics networks)
- Kanai M, Maeda Y, Okada Y. (2018) Grimon: Graphical interface to visualize multi-omics networks. Bioinformatics 34:3934-3936. [PubMed]


- github page [Link]
Softwares and data source used in Okada et al. Nature (2014)
- Okada Y, Wu D, Trynka G et al. (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376-381. [PubMed]
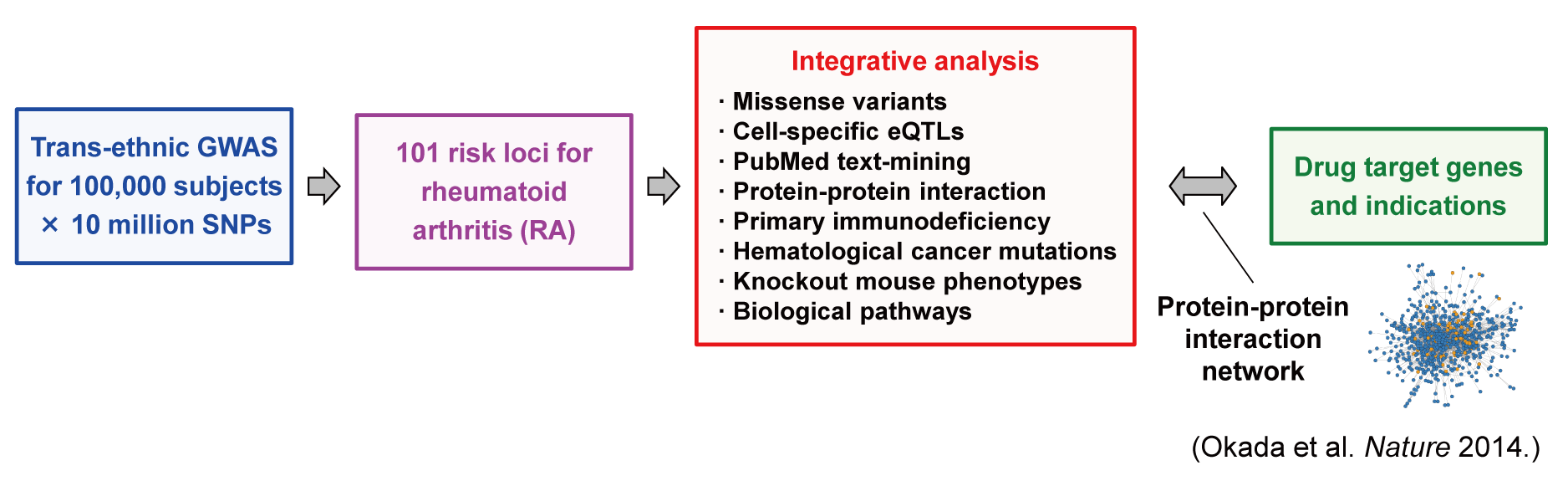
- Summary statistics of RA GWAS meta-analysis
- Trans-ethnic RA GWAS meta-analysis (19,234 RA cases and 61,565 controls) [Link]
- Eurpean RA GWAS meta-analysis (14,361 RA cases and 43,923 controls) [Link]
- Eurpean RA GWAS meta-analysis (8,875 RA cases and 29,367 controls, non-immunochip) [Link]
- Asian RA GWAS meta-analysis (4,873 RA cases and 17,642 controls) [Link]
- Summary results of RA risk SNPs in 101 risk loci [Link]
- Softwares for 1KG imputation and GWAS meta-analysis
- Perl source codes for 1KG imputation reference panel preparation [Link]
- Perl source codes for splitting GWAS data / 1KG reference panel into chuncks [Link]
- Perl source codes for handling association analysis results [Link]
- Java package for genomic control (GC) correction [Link]
- Java package for GWAS meta-analysis [Link]
- R source codes for plotting Manhattan / QQ plots of GWAS P-values [Link]
- Pleiotropy analysis using GWAS catalogue database
- Curated phenotype/SNP data from the GWAS catalogue (for data downloaded on January 31, 2013) [Link]
- H3K4me3 histone mark enrichment analysis for GWAS signals
- Software for enrichment analysis and histone mark data (from Raychaudhuri lab and the Broad Instiute) [Link]
- Softwares and data source for in-silico pipeline to prioritize biological genes from GWAS risk loci
- Java package for calculating LD between SNPs and 1KG reference data [Link]
- R source codes for assigning LD regions to SNPs [Link]
- Perl source codes for assiging UCSC genes into LD regions [Link]
- Functional annotation of SNPs (Annovar software) [Link]
- eQTL data for PBMC (from http://genenetwork.nl) [Link]
- Cell-specific eQTL data (from ImmVar project) [Link]
- PubMed text-mining (GRAIL software) [Link]
- Protein-protein interaction analysis (DAPPLE software) [Link]
- Primary immunodificiency (PID) gene list (from Journal web site) [Link]
- Cancer somatic mutation gene list (from Journal web site) [Link]
- Knockout mouse phenotype and gene list (from Journal web site) [Link]
- Molecular pathway analysis (MAGENTA software) [Link]
- Softwares and data source for GWAS-drug target overlap analysis